
What is Cystic Fibrosis? How to manage it? What are the precautions to be taken? What are the signs and symptoms? What is the cause of this disease? How to treat it? How can homeopathy help you? All of this answered, in this post and of course our doctors always there to help you. Just fill in your details in the form down below and we will answer all your questions for FREE!
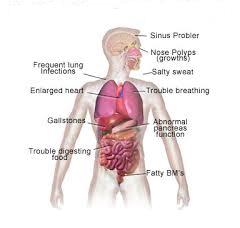
What is cystic fibrosis?
Cystic fibrosis is an inherited condition of secretory glands, including mucus and sweat-producing glands. Cystic fibrosis mostly affects the lungs, pancreas, liver, intestines, and sex organs.
In cystic fibrosis, more salt is lost during sweating. This causes imbalance of minerals in the blood and therefore causes various health issues.
In cystic fibrosis risk of diabetes, osteoporosis, infertility in men and conceiving problem in women increases.
What causes cystic fibrosis?
Cystic fibrosis is caused by a defective gene.
Children who inherit a faulty CFTR gene from their parents will be “CF carriers.”
Cystic fibrosis signs and symptoms?
Symptoms in newborns may include:
-Delayed growth
-Failure to gain weight
No bowel movements in first 24 to 48 hours after birth
Symptoms of digestive system:
-Nausea and loss of appetite
-Pale or clay-colored stool that smells foul.
-Weight loss, dehydration
-Pancreatitis
-Gallstones
-Liver diseases
-Constipation
-Diabetes
-Rectal prolapse
Symptoms of respiratory system:
-Frequent coughing
-Pseudomona infection
-Pneumothorax
Bronchiectasis
-Frequent sinusitis
-Recurrent episodes of pneumonia.
Other signs and symptoms:
-Fever
-More sputum
-Tiredness/Weakness
-Heat stroke
-Low B.P.
-Infertility
Investigations of cystic fibrosis?
certain tests include:
-Sweat chloride test is the diagnostic test as high salt level in sweat indicates cystic fibrosis.
-Chest x-ray or CT scan
-Lung function tests.
-Sputum culture
-Secretin stimulation test
-Fecal fat test
-Measurement of pancreatic function
-Trypsin and chymotrypsin in stool
Is Cystic Fibrosis curable?
No, it is not curable, but treatment helps in preventing, controlling infection and loosens mucus from the lungs.
Is Cystic Fibrosis fatal?
Cystic fibrosis(CF) is common fatal genetic disease . There is no particular cure for cystic fibrosis. Cystic fibrosis causes various effects on the body, but it mainly affects the digestive system and lungs.
Treatment of cystic fibrosis?
Treatment for lung problems includes:
-Antibiotics
Flu vaccine and pneumococcal polysaccharide vaccine yearly
-Lung transplant in some cases
-Oxygen therapy
-administer Oral pancreatic enzymes
-Anti-inflammatory medicines
Diet and management of cystic fibrosis?
-High protein diet
-Supplements of vitamins.
-High-calorie diet
-deep breathing exercise helps to loosen the mucus.
-Exercise helps improve your overall physical condition.
For more information, you can visit WiebMD and eMedicine.
We have a strong web presence all across the globe with patients in major countries like United States, Australia, United Arab Emirates, Canada, United Kingdom, most European countries, & even smaller counties like Uganda, Nepal, Bangladesh and many more.
We have a very efficient team of doctors which includes the right combination of highly experienced doctors and the doctors of the new age.
Our main aim is to make the patient comfortable so that the case can be taken with ease and the patient be treated properly.
** The text on this website is sourced from websites like emedicine and/or other verified material by government agencies around the globe along with valuable inputs and additions by our team. The content of this page is proofread and updated by the team of doctors, every once in a while, to provide the most accurate information.


